Phonon Calculation¶
To use this feature, make sure you have installed Phonopy: https://phonopy.github.io/phonopy/
[1]:
import mdapy as mp
from mdapy.phonon import Phonon
mp.__version__
[1]:
'1.0.5a2'
Build a unitcell and relax it using NEP model¶
[2]:
Al = mp.build_crystal("Al", "fcc", 4.05)
[3]:
Al.calc = mp.NEP("../../tests/input_files/UNEP-v1.txt")
[4]:
fy = mp.FIRE(Al, optimize_cell=True)
assert fy.run(100)
Step Energy fmax pressure
0 -14.965581 0.040863 -0.002461
1 -14.965630 0.038489 -0.002318
2 -14.965716 0.033869 -0.002041
3 -14.965820 0.027260 -0.001643
4 -14.965917 0.019029 -0.001148
5 -14.965986 0.009643 -0.000582
6 -14.966009 0.000360 0.000022
7 -14.966008 0.002114 -0.000128
8 -14.966008 0.002051 -0.000124
9 -14.966008 0.001957 -0.000118
10 -14.966009 0.001835 -0.000111
11 -14.966009 0.001685 -0.000102
12 -14.966009 0.001511 -0.000091
13 -14.966009 0.001314 -0.000079
14 -14.966009 0.001098 -0.000066
15 -14.966009 0.000865 -0.000052
16 -14.966009 0.000619 -0.000037
17 -14.966009 0.000365 -0.000022
18 -14.966009 0.000105 -0.000006
19 -14.966009 0.000157 0.000009
20 -14.966009 0.000091 0.000006
Converged!
Initialize a Phonon object¶
[5]:
pho = Phonon(
path="0.0 0.0 0.0 0.5 0.0 0.5 0.625 0.25 0.625 0.375 0.375 0.75 0.0 0.0 0.0 0.5 0.5 0.5",
labels="$\\Gamma$ X U K $\\Gamma$ L",
unitcell=Al,
symprec=1e-3,
)
Calculate band structure¶
[6]:
pho.compute_band_structure()
[7]:
fig, ax = mp.set_figure(figdpi=150)
fig, ax = pho.plot_band_structure(fig, ax)
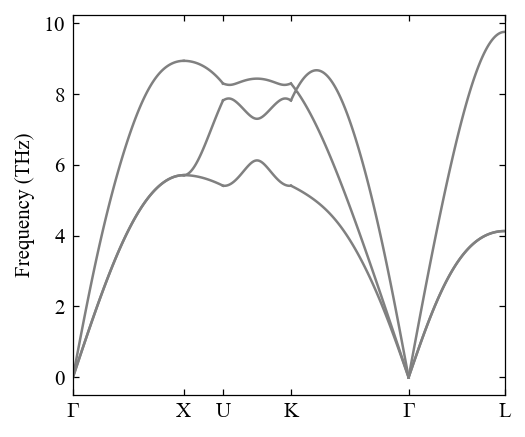
Calculate dos and pdos¶
[8]:
pho.compute_dos()
pho.compute_pdos()
[9]:
fig, ax = mp.set_figure(figdpi=150)
_ = pho.plot_dos(fig, ax)
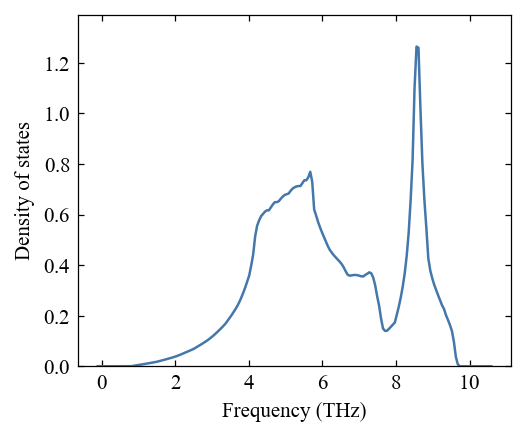
[10]:
fig, ax = mp.set_figure(figdpi=150)
_ = pho.plot_pdos(fig, ax)
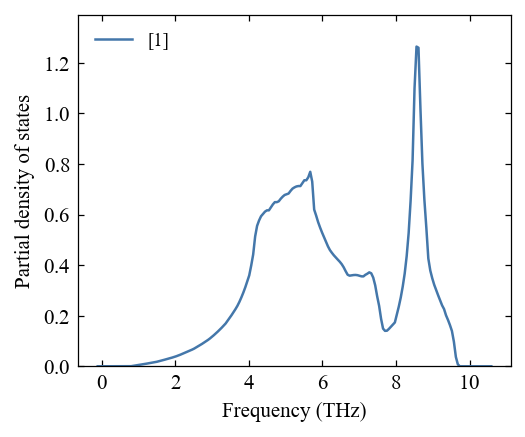
Calculate thermal properties¶
[11]:
pho.compute_thermal(300, 100, 1000)
[12]:
fig, ax = mp.set_figure(figdpi=150)
_ = pho.plot_thermal(fig, ax)
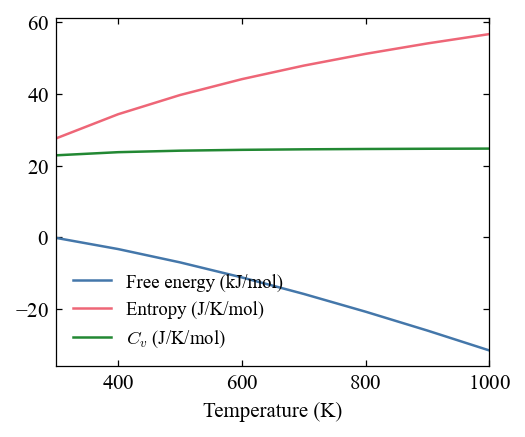
Use EAM potential¶
We first generate a EAM file: Al.eam.alloy
[13]:
_ = mp.EAMGenerator(['Al'])
[14]:
eam = mp.EAM('Al.eam.alloy')
[15]:
fig, ax = eam.plot()
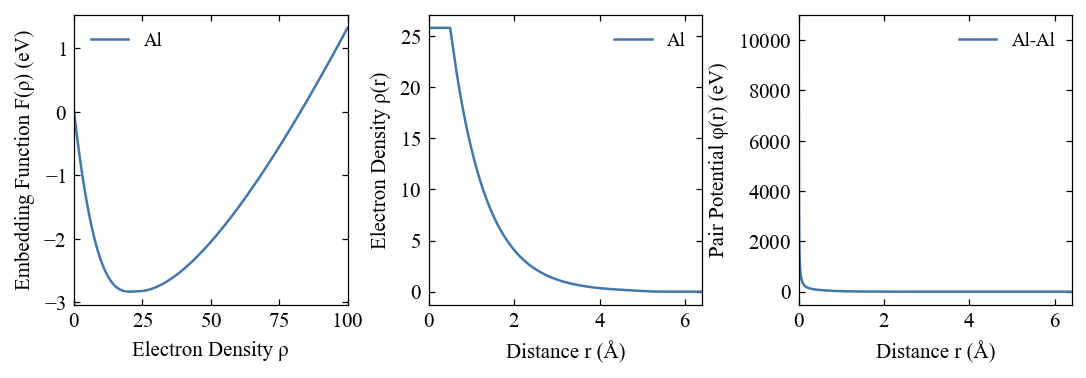
[16]:
Al = mp.build_crystal("Al", "fcc", 4.05)
Al.calc = eam
fy = mp.FIRE(Al, optimize_cell=True)
assert fy.run(100)
Step Energy fmax pressure
0 -14.320009 0.001164 0.000070
1 -14.320009 0.001096 0.000066
2 -14.320009 0.000962 0.000058
3 -14.320009 0.000772 0.000046
4 -14.320009 0.000536 0.000032
5 -14.320009 0.000268 0.000016
6 -14.319916 0.000264 0.000016
7 -14.319916 0.000035 -0.000002
Converged!
[17]:
pho = Phonon(
path="0.0 0.0 0.0 0.5 0.0 0.5 0.625 0.25 0.625 0.375 0.375 0.75 0.0 0.0 0.0 0.5 0.5 0.5",
labels="$\\Gamma$ X U K $\\Gamma$ L",
unitcell=Al,
symprec=1e-3,
)
[18]:
pho.compute_band_structure()
fig, ax = mp.set_figure(figdpi=150)
fig, ax = pho.plot_band_structure(fig, ax)
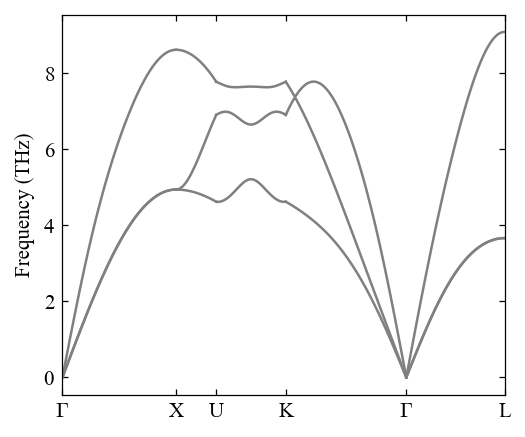
Use Lammps potential¶
To use this feature, make sure you have installed lammps python package.
[19]:
from mdapy.lammps_potential import LammpsPotential
eam = LammpsPotential(
pair_parameter="""
pair_style eam/alloy
pair_coeff * * Al.eam.alloy Al
""",
element_list=['Al']
)
[20]:
Al = mp.build_crystal("Al", "fcc", 4.05)
Al.calc = eam
fy = mp.FIRE(Al, optimize_cell=True)
assert fy.run(100)
Step Energy fmax pressure
0 -14.320009 0.001164 0.000070
1 -14.320009 0.001096 0.000066
2 -14.320009 0.000962 0.000058
3 -14.320009 0.000772 0.000046
4 -14.320009 0.000536 0.000032
5 -14.320009 0.000268 0.000016
6 -14.319916 0.000264 0.000016
7 -14.319916 0.000035 -0.000002
Converged!
[21]:
pho = Phonon(
path="0.0 0.0 0.0 0.5 0.0 0.5 0.625 0.25 0.625 0.375 0.375 0.75 0.0 0.0 0.0 0.5 0.5 0.5",
labels="$\\Gamma$ X U K $\\Gamma$ L",
unitcell=Al,
symprec=1e-3,
)
[22]:
pho.compute_band_structure()
fig, ax = mp.set_figure(figdpi=150)
fig, ax = pho.plot_band_structure(fig, ax)
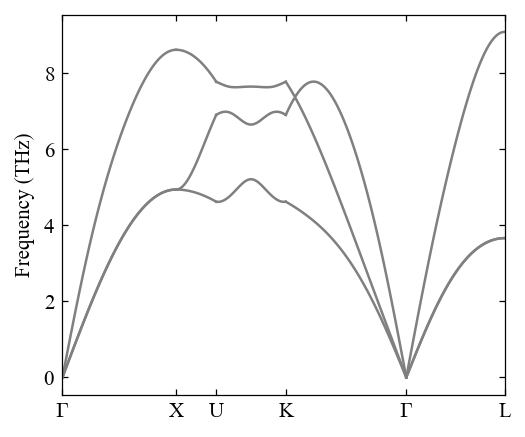
[23]:
import os
os.remove('Al.eam.alloy')