QuicklyStart
This is a quick start guide to show users what mdapy can do and how it should be implemented, for more specific information check out the API in the documentation.
Import corresponding packages
[1]:
import mdapy as mp
import numpy as np
import os
mp.init() # Run on CPU
[Taichi] version 1.6.0, llvm 15.0.1, commit f1c6fbbd, win, python 3.8.0
[Taichi] Starting on arch=x64
[2]:
mp.__version__
[2]:
'0.9.9'
Generate a System class from a dump file, which can be found in example folder and come from the Supplementary materials of this paper.
[3]:
system = mp.System('../../../example/CoCuFeNiPd-4M.dump')
Check the data of System.
[4]:
system
[4]:
Filename: ../../../example/CoCuFeNiPd-4M.dump
Atom Number: 8788
Simulation Box:
[[47.36159615 0. 0. ]
[ 0. 47.46541884 0. ]
[ 0. 0. 47.46849764]
[-1.18079807 -1.23270942 -1.23424882]]
TimeStep: 0
Boundary: [1, 1, 1]
Particle Information:
shape: (5, 5)
┌─────┬──────┬───────────┬───────────┬───────────┐
│ id ┆ type ┆ x ┆ y ┆ z │
│ --- ┆ --- ┆ --- ┆ --- ┆ --- │
│ i64 ┆ i64 ┆ f64 ┆ f64 ┆ f64 │
╞═════╪══════╪═══════════╪═══════════╪═══════════╡
│ 1 ┆ 2 ┆ 0.006118 ┆ -0.310917 ┆ -0.345241 │
│ 2 ┆ 4 ┆ 1.9019 ┆ -0.292456 ┆ 1.48488 │
│ 3 ┆ 3 ┆ -0.015641 ┆ 1.58432 ┆ 1.43129 │
│ 4 ┆ 5 ┆ 1.86237 ┆ 1.51117 ┆ -0.372278 │
│ 5 ┆ 5 ┆ 3.79257 ┆ -0.331891 ┆ -0.37583 │
└─────┴──────┴───────────┴───────────┴───────────┘
[5]:
system.data.head()
[5]:
shape: (5, 5)
| id | type | x | y | z |
|---|---|---|---|---|
| i64 | i64 | f64 | f64 | f64 |
| 1 | 2 | 0.006118 | -0.310917 | -0.345241 |
| 2 | 4 | 1.9019 | -0.292456 | 1.48488 |
| 3 | 3 | -0.015641 | 1.58432 | 1.43129 |
| 4 | 5 | 1.86237 | 1.51117 | -0.372278 |
| 5 | 5 | 3.79257 | -0.331891 | -0.37583 |
Calculate the average entropy fingerprint.
[6]:
system.cal_atomic_entropy(rc=3.6*1.4, sigma=0.2, compute_average=True, average_rc=3.6*0.9)
Calculate the CSP.
[7]:
system.cal_centro_symmetry_parameter()
Calculate the CNA pattern.
[8]:
system.cal_common_neighbor_analysis(3.6*0.8536)
Calculate the Voronoi volume.
[9]:
system.cal_voronoi_volume()
Check the calculated results.
[10]:
system.data.head()
[10]:
shape: (5, 12)
| id | type | x | y | z | atomic_entropy | ave_atomic_entropy | csp | cna | voronoi_volume | voronoi_number | cavity_radius |
|---|---|---|---|---|---|---|---|---|---|---|---|
| i64 | i64 | f64 | f64 | f64 | f64 | f64 | f64 | i32 | f64 | i32 | f64 |
| 1 | 2 | 0.006118 | -0.310917 | -0.345241 | -5.997982 | -6.469181 | 0.100696 | 1 | 12.68101 | 15 | 3.675684 |
| 2 | 4 | 1.9019 | -0.292456 | 1.48488 | -6.640986 | -6.677864 | 0.139543 | 1 | 12.012947 | 14 | 3.581766 |
| 3 | 3 | -0.015641 | 1.58432 | 1.43129 | -6.821842 | -6.666716 | 0.094929 | 1 | 12.197214 | 12 | 3.674408 |
| 4 | 5 | 1.86237 | 1.51117 | -0.372278 | -6.95832 | -6.940528 | 0.072999 | 1 | 12.900968 | 15 | 3.713117 |
| 5 | 5 | 3.79257 | -0.331891 | -0.37583 | -6.679067 | -6.846047 | 0.046358 | 1 | 12.400861 | 14 | 3.645415 |
Check the cutoff distance now.
[11]:
system.rc
[11]:
5.04
Neighbor atom index of atom 0 withing the cutoff distance.
[12]:
system.verlet_list[0][system.verlet_list[0]>-1]
[12]:
array([ 896, 8678, 897, 1009, 2, 7777, 3, 1, 110, 109, 7779,
7885, 7782, 7785, 7778, 8677, 7776, 1012, 1010, 8683, 1008, 1007,
902, 901, 899, 8785, 8787, 895, 894, 115, 113, 111, 7887,
7886, 108, 10, 9, 7, 6, 5, 4, 8676, 8786, 7890])
Corresponding distance from atom 0 to its neighbor atoms.
[13]:
system.distance_list[0][system.verlet_list[0]>-1]
[13]:
array([2.51207599, 2.57315649, 2.57907233, 2.58034816, 2.59777966,
2.60048836, 2.60123122, 2.63508509, 2.63799722, 2.64905416,
2.72325674, 2.75477768, 4.61796939, 4.39168451, 4.72094208,
4.56321812, 3.89905069, 4.59982507, 4.55429545, 4.32684219,
4.3158718 , 5.02072069, 4.39625655, 4.94515661, 4.54723002,
4.35033004, 4.48415851, 4.33703698, 3.40006632, 4.52692755,
4.59187976, 4.58003255, 4.66010003, 4.75302553, 3.73204205,
4.42655427, 3.47691914, 4.43249288, 3.67048701, 4.59684361,
3.78663373, 4.28528359, 4.63487321, 4.59095276])
Validate the distance between atom 0 and atom 896.
[14]:
system.atom_distance(0, 896)
[14]:
2.5120753608164965
Save the results to the disk.
[15]:
system.write_dump()
Do the spatial binning of entropy along xy plane.
[16]:
system.spatial_binning('xy', 'ave_atomic_entropy', 4.)
Binning coordinations.
[17]:
system.Binning.coor
[17]:
{'x': array([-0.289603, 3.710397, 7.710397, 11.710397, 15.710397, 19.710397,
23.710397, 27.710397, 31.710397, 35.710397, 39.710397, 43.710397]),
'y': array([-0.559109, 3.440891, 7.440891, 11.440891, 15.440891, 19.440891,
23.440891, 27.440891, 31.440891, 35.440891, 39.440891, 43.440891])}
Plot the binning results.
[18]:
fig, ax = system.Binning.plot(bar_label='average_entropy')
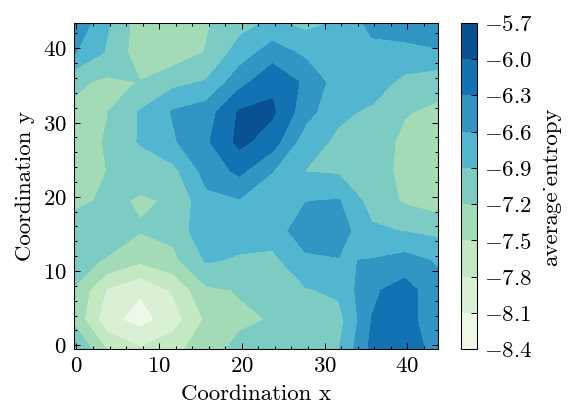
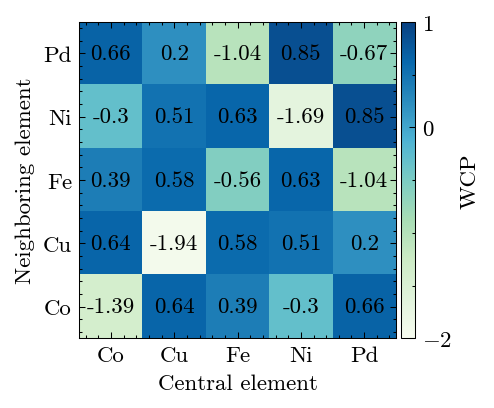
Calculate the WCP to reveal the short-range order in alloy.
[19]:
system.cal_warren_cowley_parameter()
Results show high SRO degree.
[20]:
fig, ax = system.WarrenCowleyParameter.plot(['Co', 'Cu', 'Fe', 'Ni', 'Pd'])
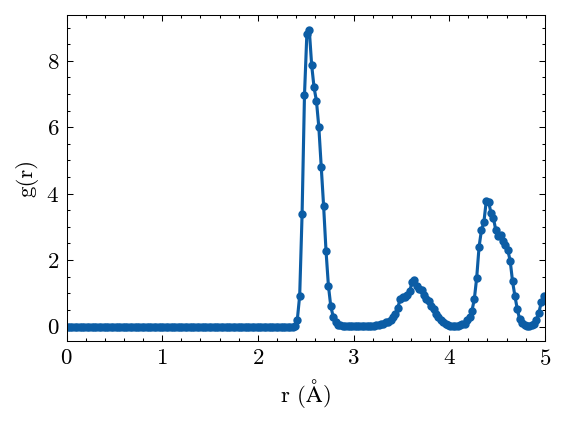
Calculate the radiul distribution function (RDF).
[21]:
system.cal_pair_distribution()
Plot the RDF results.
[22]:
fig, ax = system.PairDistribution.plot()
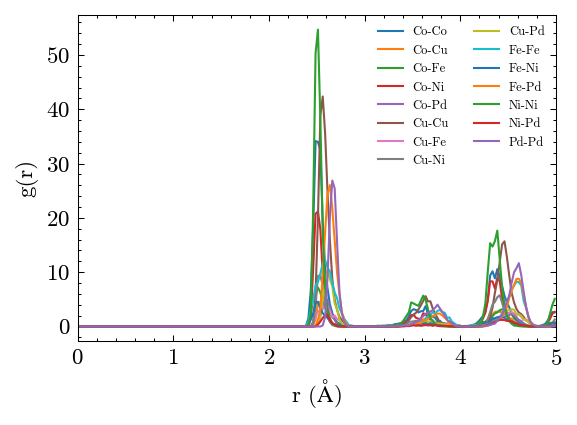
Plot the partial RDF results.
[23]:
fig, ax = system.PairDistribution.plot_partial(['Co', 'Cu', 'Fe', 'Ni', 'Pd'])
One can save the figure easily.
[24]:
fig.savefig('rdf.png', dpi=300)
os.remove('rdf.png') # Here just remove the saved figure.
Analyze the atomic trajectories
Generate a random walk trajectories.
[25]:
Nframe, Nparticles = 200, 1000
pos_list = np.cumsum(
np.random.choice([-1.0, 1.0], size=(Nframe, Nparticles, 3)), axis=0
)*np.sqrt(2)
Calculate the mean squared displacement (MSD).
[26]:
MSD = mp.MeanSquaredDisplacement(pos_list=pos_list, mode="windows")
MSD.compute()
Check the MSD results.
[27]:
MSD.msd[:10]
[27]:
array([3.39355492e-07, 5.99999611e+00, 1.20085191e+01, 1.80220932e+01,
2.40569380e+01, 3.01025624e+01, 3.61527811e+01, 4.21784838e+01,
4.81846156e+01, 5.41884617e+01])
Plot the MSD results.
[28]:
fig, ax = MSD.plot()
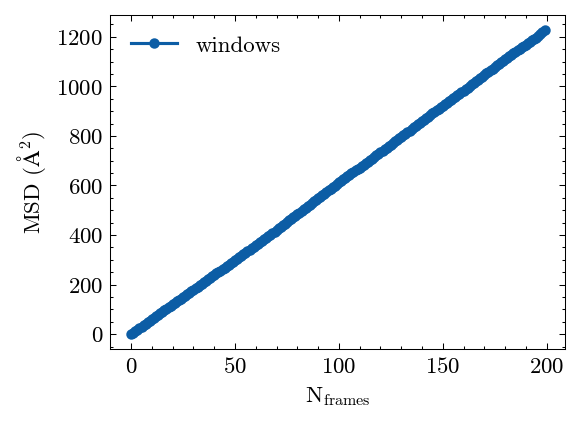
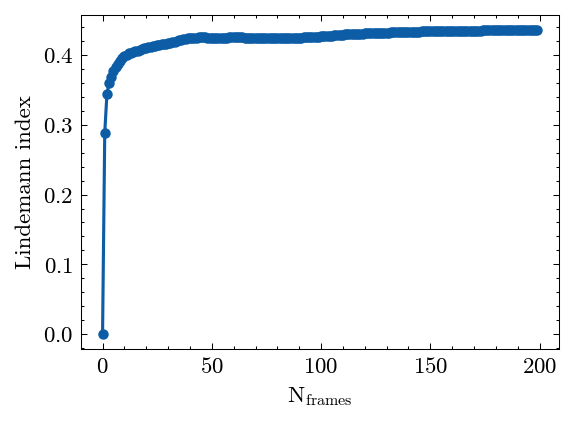
Calculate the Lindemann index.
[29]:
LDML = mp.LindemannParameter(pos_list)
LDML.compute()
Plot the Lindemann index results.
[30]:
fig, ax = LDML.plot()
Analyze EAM potential.
Generate an average EAM potential is simple.
[31]:
EAMave = mp.EAMAverage('../../../example/CoNiFeAlCu.eam.alloy', [0.2]*5)
Read this average potential file.
[32]:
potential = mp.EAM('./CoNiFeAlCu.average.eam.alloy')
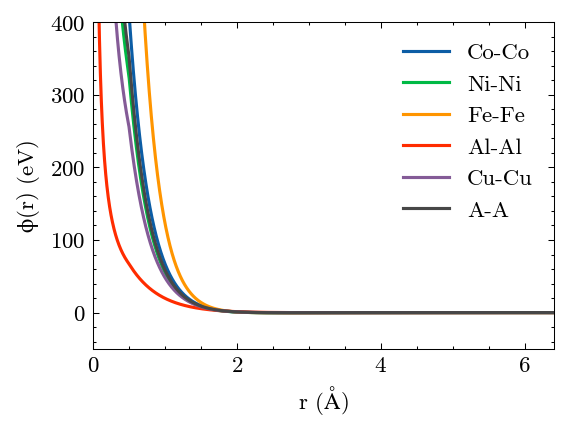
Plot the results. A is the virtual element.
[33]:
potential.plot()
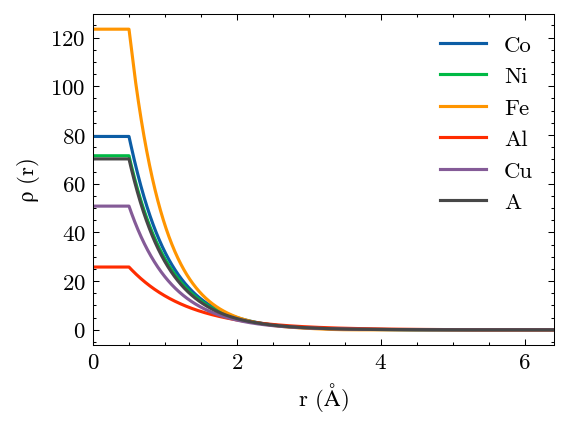
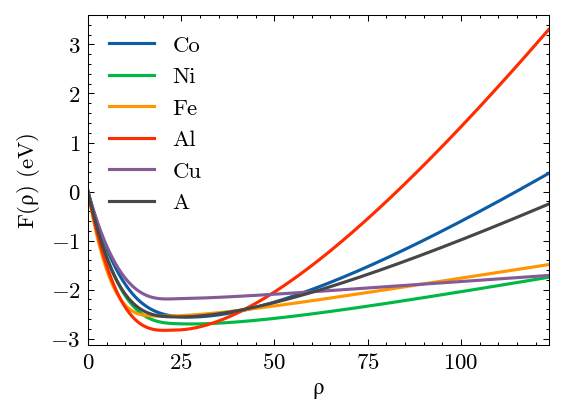
[34]:
os.remove('CoNiFeAlCu.average.eam.alloy') # Here just remove this average EAM file.